Diagnostic Techniques
 FISH
FISH
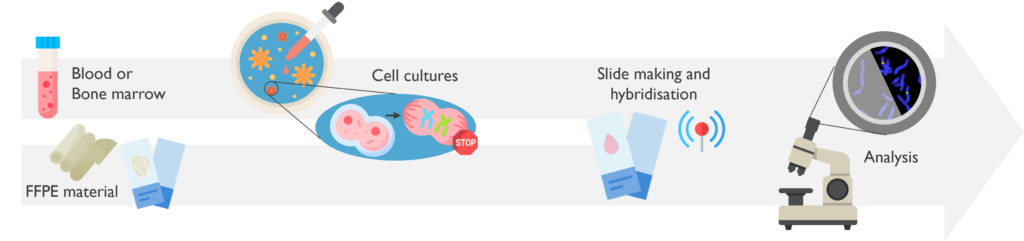
Fluorescence in situ hybridisation (FISH) is a cytogenetic technique that detects specific regions of DNA on chromosomes within cell nuclei, allowing us to locate their relative position and quantification. FISH is a rapid test which can be carried out on a wide range of sample types but only gives one result per hybridisation.
What are the sample requirements? FISH is typically performed on fixed cells from a fresh sample (such as bone marrow aspirate or peripheral blood); or formalin-fixed paraffin-embedded curls (FFPE) sections. FISH can also be carried out on fresh tumour samples and touch imprints.
If FFPE sections are sent, please send double the amount of sections for the tests required to allow for repeats. If specific regions of the sections are infiltrated with tumour, please send an accompanying H&E slide clearly marked with the tumour areas and a stated tumour content. We will use this to guide our analysis. FISH testing generally requires over 10% tumour content. Deletion testing using FISH requires 20% tumour content and can be unreliable on FFPE tissue due to signal truncation due to sectioning.
How does it work? FISH uses short DNA fluorescent sequences, complementary to the region of interest. Once the interphase cells after short-term cultures are fixed, or cells are released from FFPE sections, these fluorescent probes are hybridised (attached) in situ to the chromosomes. Finally, the slides are analysed using an epi-fluorescence microscope.
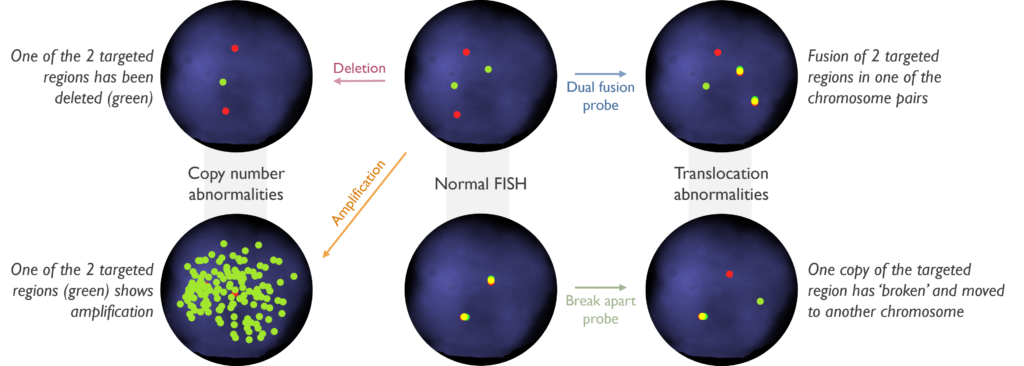
Which genetic abnormalities can it identify? FISH can be used to identify the absence of the targeted region (deletion) when fluorescence is missing. Likewise, it can detect the duplication, triplication, or amplification of the region, when more than one fluorescent signal is counted. It is also able to show when gene fusions are present by the juxta-position of specific DNA sequences, indicating translocation or other rearrangements commonly associated with malignancies.
What are the estimated turnaround times? FISH can be completed rapidly including within 24hrs in cases of urgent clinical necessity (please make sure these cases are highlighted to the laboratory for their prioritisation). All FISH is aimed to be reported within 7 days.
When is it used in Cancer testing? This technique is used, for instance, in:
- Urgent diagnosis of Acute Promyelocytic Leukaemia (APL) by the detection of PML::RARA fusion, consistent with a translocation between chromosomes 15 and 17.
- Rapid identification of gene fusions in acute leukaemia.
- Prognosis in Multiple Myeloma (MM) when there are rearrangements involving IGH.
- Risk stratification in Neuroblastoma (NB) by identifying MYCN amplification.
 Karyotyping
Karyotyping
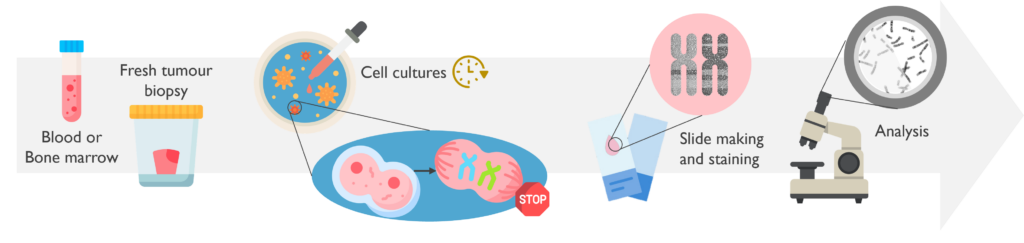
Metaphase chromosome analysis or karyotyping is a manual, visual examination of chromosomes prepared from a fresh sample that allows detection of numerical and structural chromosome anomalies.
What are the sample requirements? Samples for karyotyping must be fresh, as it required living cells, and therefore need to be received within 24 hours of sampling. The most common specimens are bone marrow aspirate and peripheral blood samples.
How does it work? Cells in the sample are grown in culture for 1-2 days. Cultures are then ‘harvested’ which arrests the dividing cells in metaphase, which is the stage in mitosis when all chromosomes become visible. Chromosome preparations are then dropped on to a slide, aged, treated with the enzyme trypsin and stained with DNA dye. This gives the DNA a reproducible banding pattern which can be recognised by trained chromosome analysts. Metaphase chromosome analysis is a complex test, that takes several days to prepare the material for, with the analysis taking several hours by skilled analysts.
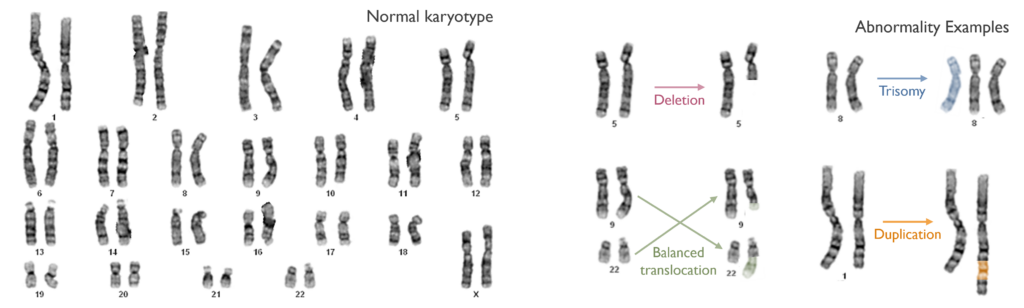
Which genetic abnormalities can it identify? Karyotyping can detect a wide variety of chromosome anomalies including changes in the number of chromosomes, structural rearrangements (e.g. deletions, duplications, inversions or translocations). It can also identify the overall genomic complexity of a sample. The resolution of karyotyping is limited though with the smallest abnormality detectable being over 5 million base pairs of DNA. Small variants (aka mutations) and small structural rearrangements might not be detected. Furthermore, as karyotyping only looks at dividing cells, if cancer cells are not dividing in culture, abnormalities may be missed.
What are the estimated turnaround times? Karyotyping studies can be completed with 72hrs in cases with urgent clinical necessity (please make sure these cases are highlighted to the laboratory for their prioritisation). Non-urgent karyotyping is aimed to be reported within 21 days of sample receipt. If karyotype studies fail to yield metaphases we will attempt SNP array (see below) in all cases, which may lead to a delay in reporting results.
When is it used in Cancer testing? This technique is used, for instance, in:
- Risk stratification in Acute Myeloid Leukaemia (AML), Primary Myelofibrosis (PMF) and Chronic Myelomonocytic Leukaemia (CMML).
- Diagnosis of Chronic Myeloid Leukaemia (CML) by detecting a balanced translocation between chromosomes 9 and 22 (Philadelphia chromosome).
- Diagnosis and prognosis in Myelodysplastic Syndrome (MDS) by assessing genomic complexity and other findings.
 SNP array
SNP array
Single Nucleotide Polymorphism (SNP) Array is a molecular test capable of identifying genome wide copy number changes and loss of heterozygosity (LOH).
What are the sample requirements? SNP Array analysis is performed on DNA. This can be extracted from any tissue type, such as peripheral blood, bone marrow or fresh tumour biopsies. DNA extracted from FFPE material can lead to poor quality results using SNP-array. We may choose to test using MethylEPIC Array in certain circumstances as this test performs better using DNA from FFPE material. SNP Array test requires at least 20% abnormal cell content, therefore a normal result from tissue with lower tumour content may be uninformative.
How does it work? The basis of this test are SNPs, which are DNA positions which vary between individuals. Once the DNA has been extracted from the tumour tissue, these SNPs are amplified and labelled with fluorescent beads (different colour for different base change) on an Illumina Infinium CytoSNP-850k BeadChipTM. Then the hybridisation signals from the microchip are scanned using an imagining system and the data is analysed using BlueFuseTM software.
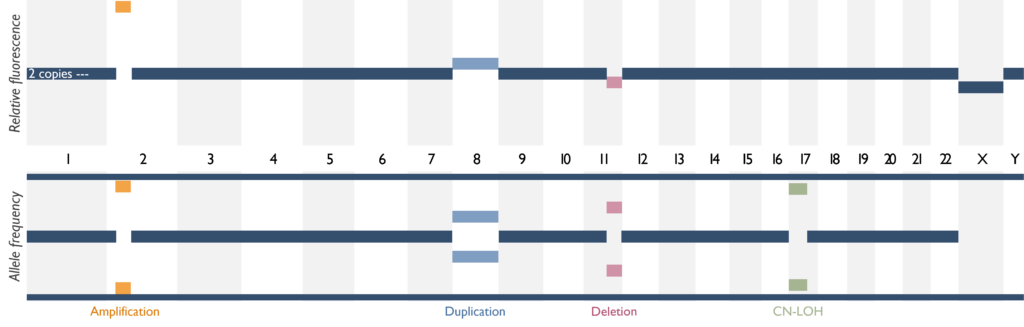
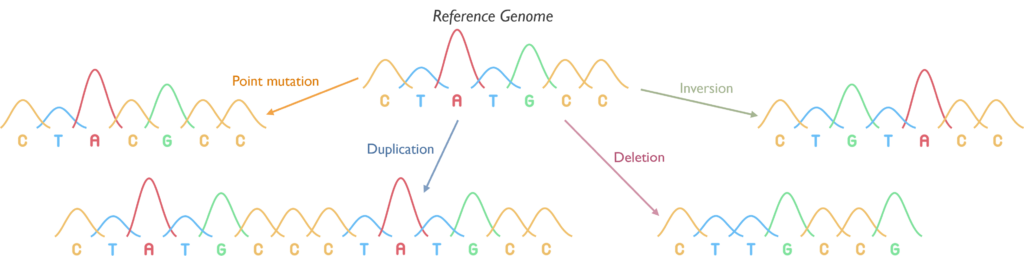
Which genetic abnormalities can it identify? SNP Array is able to identify copy number variants, i.e. gains (duplications, amplification) and losses (deletions), but also LOH, which is the loss of one of the parental copies of DNA. The image below shows how these different copy number findings appear using SNP-array. Copy neutral LOH (CN-LOH) can be equivalent to a deletion in certain scenarios, but is also relevant for regions containing a mutated gene, as it means that the mutation is likely present in both copies of the gene (known as homozygous or biallelic). Balanced rearrangements cannot be detected using this assay. This means gene fusions cannot be detected unless it indicates a deletion or duplication of a region between two key genes. Single nucleotide variants (SNVs) also cannot be detected and copy number changes below 5Mb and CN-LOH below 10Mb, in the absence of disease-relevant genes, will not be reported.
What are the estimated turnaround times? The overall TAT for SNP Array is 14-21 days, depending on the clinical urgency of the case.
When is it used in Cancer testing? This technique is used when Karyotyping fails, but also as the main test for certain malignancies, for instance, in:
- Diagnosis of Lymphoblastic Leukaemia (B-ALL) identifying key ploidy subtypes.
- Prognosis in Neuroblastoma (NB) by the detection of segmental abnormalities, whole chromosome abnormalities or MYCN amplification.
- Risk stratification in Chronic Lymphocytic Leukaemia (CLL), by the detection of trisomy 12 or deletion of 11q arm.
 MethylEPIC Array
MethylEPIC Array
The Methylation EPIC Array is able to quantify methylation by targeting significant sites across the whole genome for the detection of gains and losses of DNA material.
What are the sample requirements? This test has only been validated in FFPE samples (5 x10µM curls required) or fresh frozen tumour biopsies.
How does it work? The methodology of this assay is similar to that of the SNP Array. The main difference is that the DNA extracted from the tumour material undergoes a bisulphite conversion before being amplified. This step allows the distinction between methylated and unmethylated cytosines by converting the latter into uracils. After this step, the DNA is amplified by PCR and labelled with fluorescent beads on an Illumina Infinium MethylationEPIC BeadChipTM. Then the hybridisation signals from the microchip are scanned, using an imagining system and the data is processed via Molecular Neuropathology tool, developed by the University Hospital Heidelberg*. This platform will return a report containing the classification of the tumour based on its methylation pattern, the methylation status of the MGMT promoter (when relevant), and a low-resolution plot for copy number variants (CNVs).
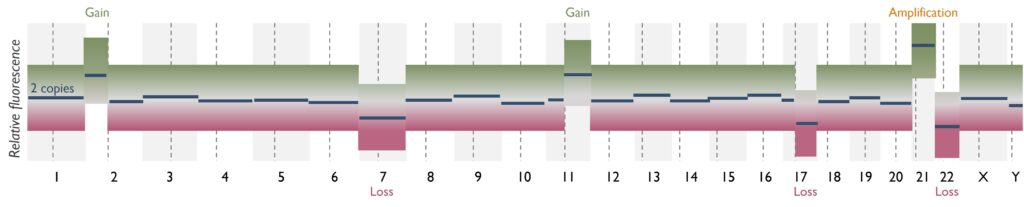
Which genetic abnormalities can it identify? MethylEPIC Array will only identify CNVs, such as gains (duplications, amplification) and losses (deletions), at a lower resolution than SNP Array. Single nucleotide variants (SNVs) and gene fusions cannot be detected using this assay.
What are the estimated turnaround times? The aimed TAT for MethylEPIC Array is 14 days from sample receipt, although it can be processed more urgently in cases of clinical necessity.
When is it used in Cancer testing? This technique is used, for instance, in:
- Aiding diagnosis* of brain tumour types, by scoring a probable classification based on the methylation profile.
- Aiding diagnosis* of atypical melanocytic lesions, in conjunction with histopathologic findings.
- Prognosis in different types of brain tumours, by determining the MGMT promoter methylation status and CNVs profile.
*Please note that classification using methylation profiling is a tool for research use only, it is not verified and has not been clinically validated and, therefore, must not be used for diagnostic purposes.
 Real-time Quantitative PCR (RQ-PCR)
Real-time Quantitative PCR (RQ-PCR)
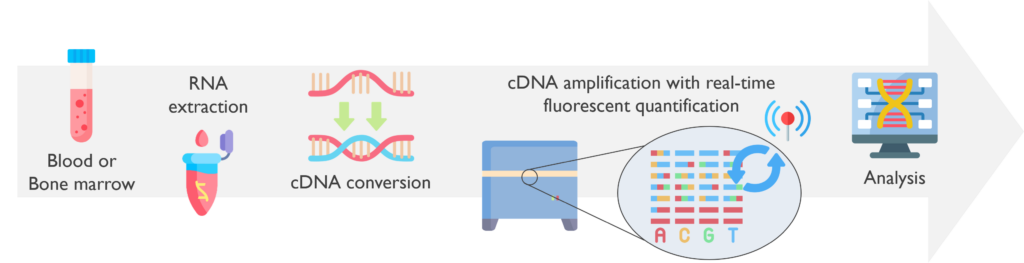
The Real-time Quantitative PCR (RQ-PCR) assay is a rapid and sensitive technique that allows the simultaneous amplification and quantification of DNA.
What are the sample requirements? This test is performed on EDTA peripheral blood samples, or bone marrow biopsies, received within 48h of being taken.
How does it work? The sample received must be processed for RNA extraction, which is then converted into coding DNA (cDNA) by a reverse transcriptase PCR (RT-PCR). On this cDNA, the RQ-PCR is performed adding an extra specific probe that targets the fusion. If it is present, it will bind it and release fluorescence when cleaved by the polymerase along the PCR cycles. The fluorescent signals are detected in real-time by a LightCycler 480. Graphs are generated from these signals to be analysed by specific software.
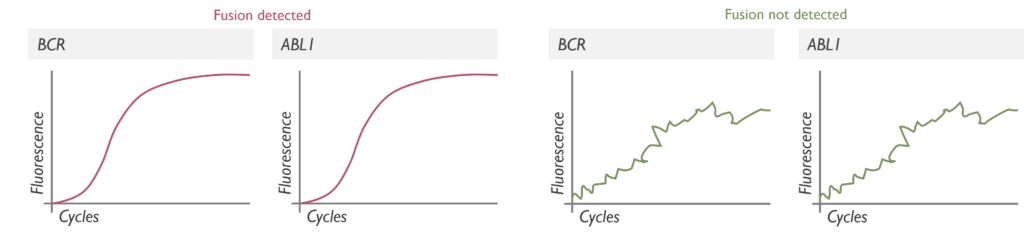
Which genetic abnormalities can it identify? The RQ-PCR is used to detect targeted gene fusions. In our lab, it is exclusively used to detect BCR–ABL1 fusion in new diagnoses of Chronic Myeloid Leukaemia (CML). It targets the most common breakpoints (e13a2 and e14a2), which cover 98% of BCR–ABL1 fusions seen in CML. If testing of the minor breakpoint transcript is required, the sample is exported to the Leeds laboratory for processing. As there is a risk of false negative results, our service generally combines findings from RQ-PCR and FISH testing.
What are the estimated turnaround times? The BCR–ABL1 test requests are usually issued within 3-7 days of sample receipt, although the turnaround time is 14 days.
When is it used in Cancer testing? This technique is used in our laboratory specifically to confirm a FISH diagnosis of BCR::ABL1 in CML (at the major breakpoint), and posteriorly to monitor its treatment with tyrosine kinase inhibitors (TKIs).
 Droplet Digital PCR (ddPCR)
Droplet Digital PCR (ddPCR)
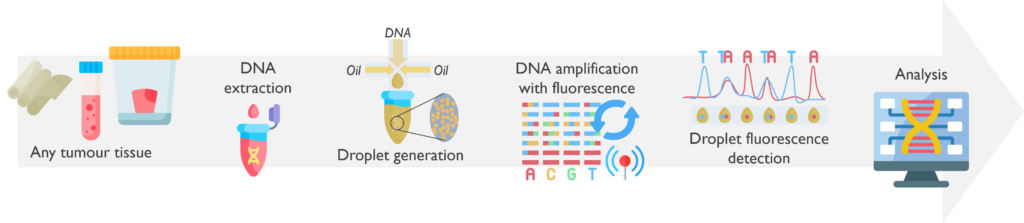
Droplet Digital PCR (ddPCR) is a highly sensitive and precise technique for detecting and quantifying targeted mutations, especially at low frequencies.
What are the sample requirements? ddPCR can be performed on DNA extracted from various types of tissue, depending on the available material and the purpose of the test, such as peripheral blood, bone marrow, FFPE samples or fresh tumour biopsies.
How does it work? The DNA is extracted from the tumour tissue received. With the BioRad Droplet Generator, samples are partitioned into nano droplets. Once droplets have been generated for all samples in the experiment, the sample plate is transferred to a thermocycler that will amplify the DNA by PCR using fluorescent probes. The products are then processed by BioRad Droplet Reader, which detects the fluorescent signal from every droplet. Finally, these results are analysed using specific software.
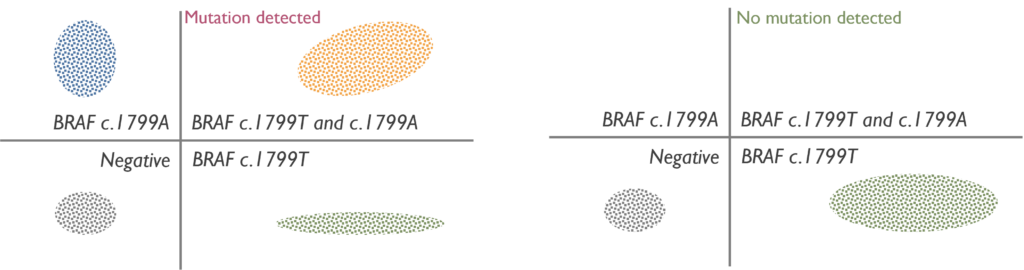
Which genetic abnormalities can it identify? While ddPCR can identify both single nucleotide variants and copy-number variants, in our Cancer laboratory is exclusively used to detect a single nucleotide variant.
What are the estimated turnaround times? The aimed TAT of ddPCR testing is 14 days, although it can be processed more urgently in cases of clinical necessity.
When is it used in Cancer testing? This technique is mainly used for the diagnosis and monitoring of Histiocytic disorders, such as Langerhans cell histiocytosis or Erdheim-Chester Disease, by detecting the most common activating variant, which is BRAF c.1799T>A.
 Sanger Sequencing
Sanger Sequencing
Sanger sequencing is a conventional DNA sequencing method that has largely been superseded by more advanced techniques like next generation sequencing (NGS), which offer higher throughput and broader applications.
What are the sample requirements? This method can be performed on DNA extracted from various types of tissue, depending on the available material and the purpose of the test, although fresh tumour samples are preferable. A minimum of 40% tumour content is required to reliably detect variants present at a minimum of 20% allele frequency.
How does it work? The DNA is extracted from the tumour sample and amplified by PCR with fluorescent chain-terminating nucleotides. This produces fragments of varying lengths, which are then processed by capillary electrophoresis with fluorescent detection. The DNA sequence is reconstructed to be analysed by specific software.
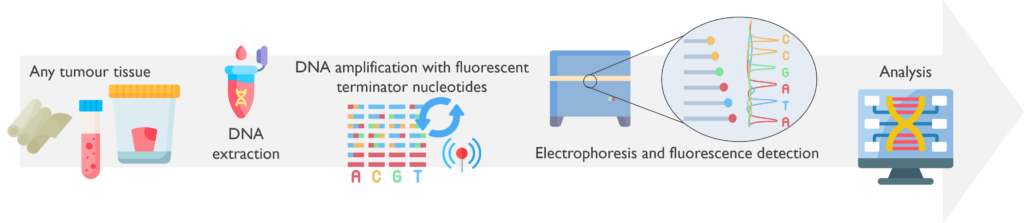
Which genetic abnormalities can it identify? Sanger sequencing is able to identify single nucleotide variants (SNVs), inversions, and small insertions, deletions or duplications in the targeted region.
What are the estimated turnaround times? The TAT of Sanger sequencing depends on the clinical urgency of the case, although is usually aimed at 7-14 days.
When is it used in Cancer testing? This technique is rarely used nowadays in our Cancer Service, although there are still a few cases when Sanger is performed:
- Diagnosis of WNT-driven Medulloblastomas (MB), which represent around 8% of them and have a relatively good prognosis. Testing of exon 3 of CTNNB1 is now only perform sporadically in certain urgent MB.
- Diagnosis of rhabdoid tumours by detecting driver-mutations in SMARCB1, associated with a poor prognosis.
 Targeted PCR fragment analysis
Targeted PCR fragment analysis
PCR Fragment analysis identifies targeted genetic abnormalities by sizing the DNA fragments following PCR.
What are the sample requirements? This method uses DNA extracted from all types of tissue, depending on the purpose of the test. The minimum tumour content required to detect a 10% allele frequency is 20%.
How does it work? The DNA is amplified by PCR using fluorescent probes. The products are then loaded into the ABI analyser for capillary electrophoresis separation where the DNA fragment sizes are called using a dedicated sensor. Finally, results are analysed using specific software. For methylation testing, a pre-PCR step of bisulphite conversion takes place to allow the distinction between methylated and unmethylated cytosines by converting the latter into uracils.
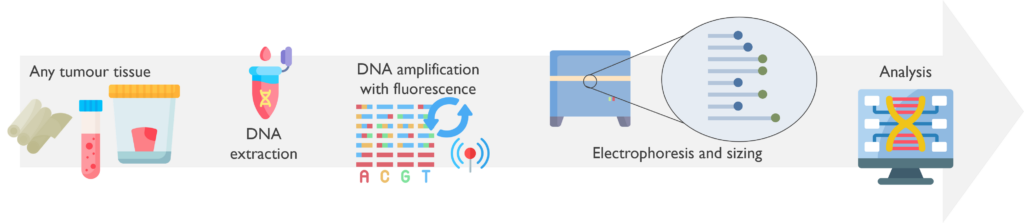
Which genetic abnormalities can it identify? PCR Fragment Analysis only assesses the size of the products and therefore detects small gains, duplications and losses. By using base-specific primers of different sizes, it can also detect methylation status or single nucleotide variants. It is currently used in our Cancer service for the detection of methylation in the MGMT promoter, internal tandem duplications (ITD) in exons 14-15 of the FLT3 gene and 4 base pair insertions in exon 12 of the NPM1 gene. Please note that the sensitivity of our FLT3 assay for detecting extremely large ITDs (>213 bp) is uncertain.

What are the estimated turnaround times? The aimed TAT of Fragment analysis is currently 7 days, although it can be processed more urgently in cases of clinical necessity.
When is it used in Cancer testing? This technique is used in:
- AML testing pathway for the rapid detection of FLT3-ITD and NPM1 4bp insertions to support diagnosis and risk-stratification.
- Prognosis in in Glioblastomas (GB) by assessing the MGMT-promoter methylation status in cases where EPIC array is not successful.
 Agena MassARRAY
Agena MassARRAY
The Agena MassARRAY is a rapid, targeted molecular test able to identify targeted DNA variants in a sensitive, high throughput and cost-effective manner.
What are the sample requirements? MassARRAY works with DNA extracted from various types of tissue, depending on the available material and the purpose of the test, such as peripheral blood, bone marrow, FFPE samples or fresh tumour biopsies.
How does it work? The region of interest for each DNA is initially amplified by PCR. Then an iPLEX extension reaction takes place to produce DNA fragments of variable length using mass-modified terminator nucleotides. The resulting products are finally processed by MALDI-TOF mass spectrometry technology and analysed with MassARRAY Typer software.
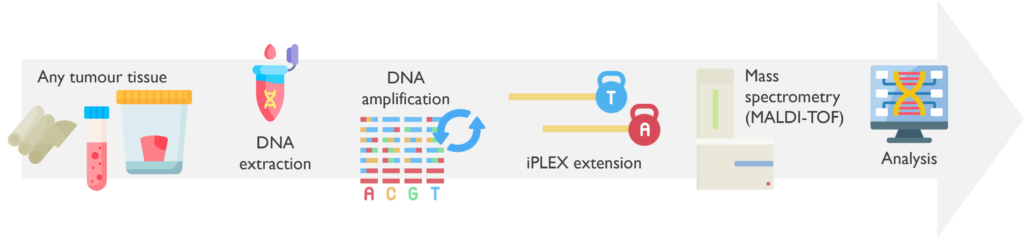
Which genetic abnormalities can it identify? This assay is limited to the detection of a single nucleotide change or deletion at a targeted position. Agena will not reliably detect variants below 5% allele frequency, which is approximately equivalent to a 10% tumour content.
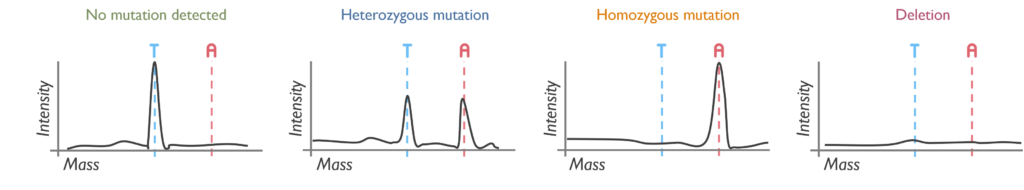
What are the estimated turnaround times? Testing by Agena has an aimed TAT of 7 days, although it can be processed more urgently in cases of clinical necessity (such as FLT3-TKD in AML or ALK in Neuroblastoma).
When is it used in Cancer testing? This technique is used, for instance, in:
- Diagnosis of Hairy Cell Leukaemia (HCL) by detecting the BRAF variant V600E in blood or bone marrow.
- Prognosis in Melanoma by testing activating variants in BRAF and NRAS genes, which are predictive biomarkers for BRAF and MEK-targeted therapies.
- Risk stratification in Neuroblastoma (NB) assessing the presence of common ALK activating variants.
- Pharmacogenetic screening in solid tumours by DPYD gene testing, to evaluate the toxicity of chemotherapy and dosage recommendation. Please note that DPYD testing is currently performed by Newcastle but reported by the Sheffield Laboratory.
Please note that this test is not accredited by ISO:15189.
 MSI-Plus Assay
MSI-Plus Assay
The MSI-Plus Assay is a rapid molecular test, mainly used in Colorectal cancer, that detects MicroSatellite Instability (MSI), BRAF V600E and activating RAS variants for the screening of patients at risk of Lynch Syndrome and recommendation of targeted therapies.
What are the sample requirements? The MSI-Plus Assay is performed on DNA extracted from 3 x 10µM FFPE curls. A minimum of 10% tumour content is required to ensure that results represent the neoplastic clone.
How does it work? The extracted DNA is amplified by multiplex PCR, using 21 primer pairs to target relevant regions. These regions cover highly specific mononucleotide repeats (MNR) used for the evaluation of MSI and common variants in BRAF, KRAS and NRAS. With the amplified targets, libraries are created and sequenced by NGS. Finally, the data is processed through a bioinformatic pipeline and results are analysed on the MSI-App.
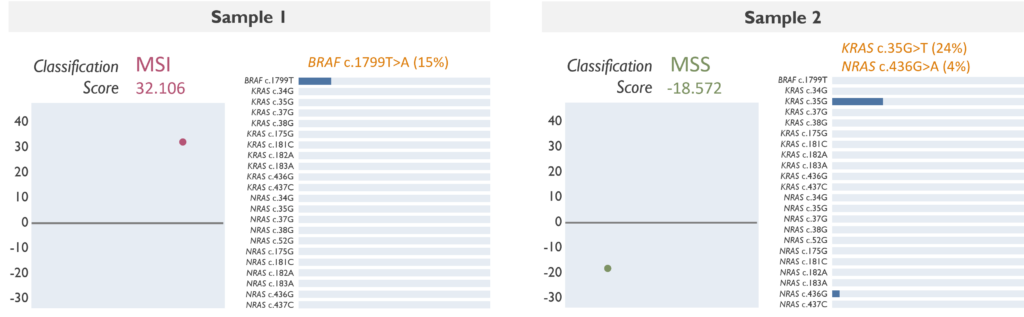
Which genetic abnormalities can it identify? This assay evaluates MSI by assigning a score based on the hypermutability of the targeted MNRs. A positive score suggests a high MS instability (MSI), whereas a negative score is indicative of MS stability (MSS). In addition, it is able to detect some of the most common variants in BRAF, KRAS and NRAS. The MSI Assay may not reliably detect variants below 5% allele frequency, although any targeted variant with an allele frequency below 4% will not be reported.
What are the estimated turnaround times? The MSI-Plus Assay has a quick TAT aimed at 7 days.
When is it used in Cancer testing? This technique is used to:
- Identify CRC patients at risk of having Lynch Syndrome when MSI is detected in the absence of BRAF V600E.
- Predict the response of CRC patients when treated with targeted treatments, such as anti-EGFR therapy.
- Assess the eligibility of patients, both with CRC and upper tract gastrointestinal tumours (UGI), to be treated with immune checkpoint inhibitors. Please note that for any UGI patients, only the MSI status will be included on the report, as the relevance of BRAF, KRAS or NRAS is not determined.
 NGS Gene Panels
NGS Gene Panels
A Next-Generation Sequencing (NGS) panel simultaneously targets various genes of interest for a particular disease. The genes are entirely sequenced, allowing the detection of key variants that are relevant for the clinical management of the patient.
What are the sample requirements? The NGS gene panels use DNA extracted from various types of tissue, depending on the available material and the purpose of the test, such as peripheral blood, bone marrow, FFPE samples or fresh tumour biopsies.
How does it work? The DNA is extracted from the tumour tissue available. Then the targeted genes on a particular panel are amplified by PCR with numerous primers, generating numerous fragments of all the regions of interest. The amplified DNAs are then combined into a library and sequenced by NGS. The data files generated are mapped against a reference genome, which allows detection of any variants in the sample. Finally, the possible disease-causing variants are filtered by a specific bioinformatic pipeline and classified by scientists to assess their clinical relevance.
Which genetic abnormalities can it identify? These assays can detect small somatic variants within the targeted genes included on each panel. The filtered variants can be classified in Passengers (not causing disease), VUS (Variants of Unknown Significance) or Drivers (Disease-causing variants). Driver variants are considered to be relevant, and therefore included on the report to the clinician.
What are the estimated turnaround times? The aimed TAT on NGS panels is 21 days. Work is underway to improve reporting times for NGS panels to within 14 days.
When is it used in Cancer testing? There are numerous gene panels available to help with the diagnosis, prognosis and risk stratification of many types of cancer, for instance:
- Paediatric Oncology Panel, for Paediatric tumours such as Neuroblastoma, Medulloblastoma, Rhabdoid tumours.
- Histiocytosis Panel, for histiocytic disorders.
- Other panels, like the TruSight Oncology 500 (TSO500), PanHaemOnc (PHO) or MPN Panels, are tested in the YNE-GLH laboratory in Leeds and reported by either Leeds or Newcastle service.
 RNA Fusion NGS Panel
RNA Fusion NGS Panel
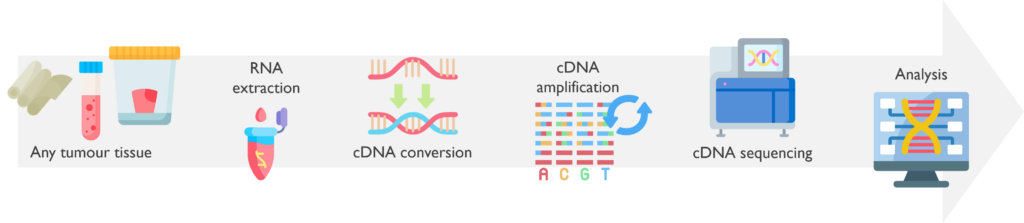
The Illumina TruSight RNA Fusion Panel is another NGS-based test, which uses RNA extracted from a tumour sample to identify possible fusions between relevant somatic genes.
What are the sample requirements? RNA can be extracted from FFPE (5 x10 µM curls), fresh, or frozen tumour biopsies, depending on the type of tumour. This assay has been validated down to a sensitivity of 2% tumour content.
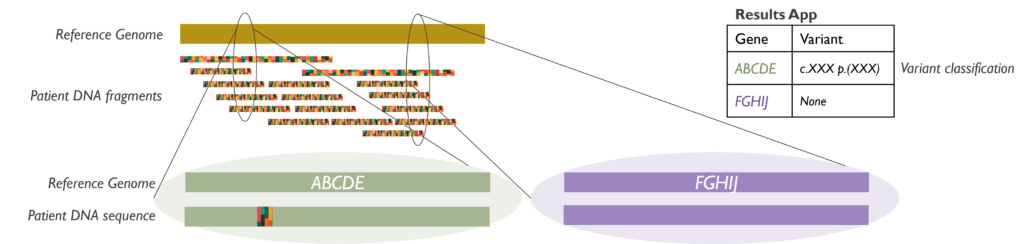
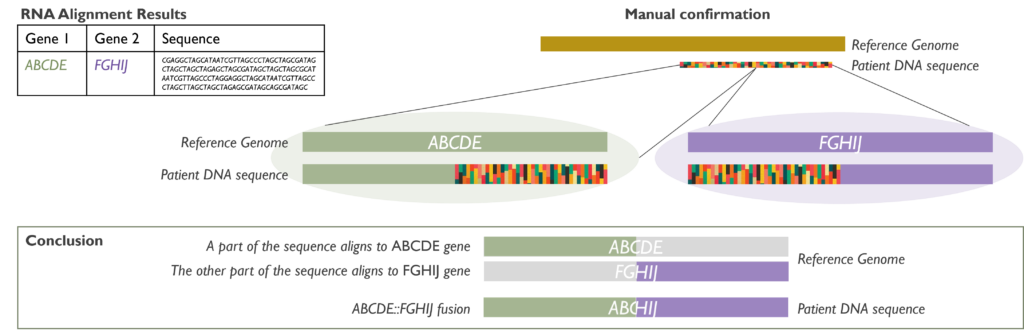
How does it work? It is crucial that RNA (and not DNA) is extracted from the samples received. Once the RNA is available, it is converted to double-stranded coding DNA (cDNA) and amplified. The amplicons are then pooled into libraries that are sequenced by NGS. Data files are processed through an RNA alignment bioinformatic pipeline and results are analysed following various steps to confirm the results found and their relevance to each case.
Which genetic abnormalities can it identify? The RNA fusion panel is able to detect fusions involving at least one of 507 clinically relevant cancer genes and identifying its partner gene, either within the established gene list or as a novel fusion.
What are the estimated turnaround times? RNA fusion panel testing is aimed to be reported before 21 days if activated at sample receipt, although it can be processed more urgently in cases of clinical necessity. Please note that many cases will exceed 21 days, as RNA fusion may be used to confirm findings from other techniques, such as FISH, or as a last resource in certain tumours if no stratifying risks have been found in previous tests.
When is it used in Cancer testing? This technique can be used to confirm results, such as FISH rearrangements with an unknown partner, or as the main testing in certain malignancies. For instance, in:
- Diagnosis of a specific subtype of sarcoma or neurological tumour, such as JAZF1::SUZ12 in endometrial stromal sarcoma.
- Identification of patients eligible for tyrosine kinase inhibitor therapy e.g. NTRK inhibitors in various types of tumour.
- Risk stratification in B-cell precursor Acute Lymphoblastic Leukaemia (B-ALL) when no other findings are available.
 Whole genome sequencing (WGS)
Whole genome sequencing (WGS)
Whole genome sequencing (WGS) provides a comprehensive genome-wide view of the known genetic alterations that drive development and progression of tumours. WGS is now funded by NHS England and available for a subset of eligible patients with solid tumours and haematological malignancies.
What are the sample requirements? This technique usually uses DNA extracted from fresh tumour biopsies and a matching peripheral blood sample to compare both somatic and germline mutations. The somatic variant detection will be significantly reduced for tumour contents below 30%.
How does it work? Both fresh tumour and germline samples are usually sent from the Newcastle laboratory to the central NEYGLH Leeds laboratory for DNA extraction, except Medulloblastomas (MBs) and Neuroblastomas (NBs) that are extracted in-house. Leeds will then coordinate the export of both DNAs to the Sequencing Centre at the Wellcome Genome Campus in Cambridge for WGS testing. Post-sequencing data will then be processed by a specific bioinformatic pipeline and uploaded to an interpretation portal. Cancer Clinical Scientists from Newcastle will finally analyse, interpret and report any WGS findings.
In all cases, WGS results will be discussed at the Genomics Tumour Advisory Board (GTAB) meetings before writing a final report. Haematological reports will be issued via Specialist Integrated Haematological Diagnostic Service (SIHMDS) and solid tumour reports will be integrated into the appropriate histopathology system.
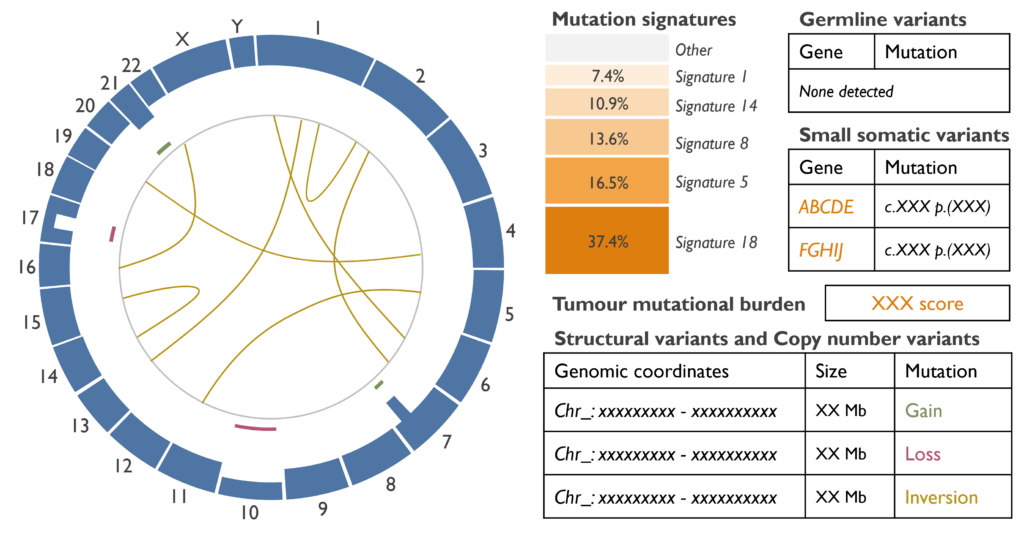
Which genetic abnormalities can it identify? By sequencing the whole genome, WGS can identify nearly all types of mutations, which are small somatic variants (SSV), such as single nucleotide variants (SNVs) and small indels; and copy number and structural variants (CNVs and SVs), such as gains, losses, loss of heterozygosity (LOH), translocations or inversions. Any germline variant that may be present and relevant will also be presented for analysis.
There are two additional concepts as well, which are the Tumour mutational burden (TMB) and the mutation signatures. The TMB indicates the total number of mutations present in the DNA, which can be useful for treatment guidance (i.e. high TMB tumours could benefit from immunotherapy). The mutation signatures
What are the estimated turnaround times? WGS testing in Cancer Services has an intended TAT of 84 days from activation, although it can be processed more urgently in cases of clinical necessity.
When is it used in Cancer testing? WGS is usually only triggered when all the main standard testing (aka Standard of Care, SOC) has been exhausted with no diagnosis or useful findings for clinical management. Cases for submission will be determined via discussion with the WGS leads and the clinicians in their regions. For instance, WGS can be performed in:
- Sub-typing sarcomas or neurological tumours when other SOC did not have a clear outcome.
- Characterising paediatric cancers when existing testing has not identified a clear genetic abnormality.
- Risk stratification in Neuroblastomas when all other SOC are uninformative.